

Published on 20.09.2017
| Table 1: Revised 2010 Task force criteria, modified from Marcus et al. [16]. | ||
| I. Global or regional dysfunction/structural alterations | ||
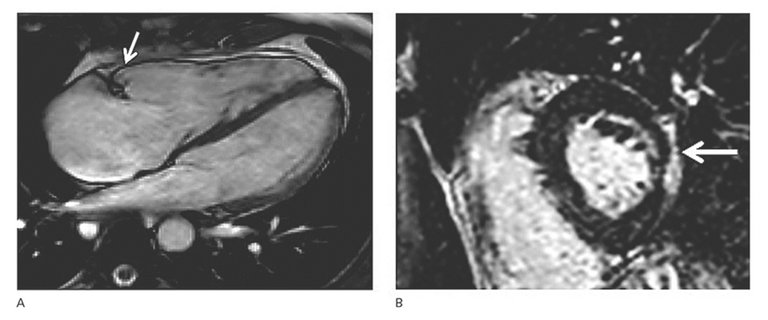
| Major | 2D transthoracic echocardiography | • Regional RV akinesia, dyskinesia, or aneurysm •And 1 of the following criteria (end diastole): – Parasternal Long Axis (PLAX) RVOT ≥32 mm (PLAX/BSA ≥19 mm/m2) – Parasternal Short Axis (PSAX) RVOT ≥36 mm (PSAX/BSA ≥21 mm/m2) – or RV fractional area change ≤33% |
| Cardiac magnetic resonance tomography | • Regional RV akinesia, dyskinesia, or dyssynchronous RV contraction •And 1 of the following criteria (end diastole): – RV end-diastolic volume /BSA ≥110 ml/m2 (♂) or ≥100 ml/m2 (♀) – or RV ejection fraction ≤40% | |
| RV angiography | • Regional RV akinesia, dyskinesia, or aneurysm | |
| Minor | 2D transthoracic echocardiography | • Regional RV akinesia, or dyskinesia •And 1 of the following criteria (end diastole): – PLAX RVOT ≥29–31mm (PLAX/BSA ≥16–18 mm/m2) – PSAX RVOT ≥32–35 mm (PSAX/BSA ≥18–20 mm/m2) – RV fractional area change >33 – ≤40% |
| Cardiac magnetic resonance tomography | • Regional RV akinesia, dyskinesia, or dyssynchronous RV contraction •And 1 of the following criteria (end diastolic): – RV end-diastolic volume/BSA ≥100–109 ml/m2 (♂) or ≥90–99 ml/m2 (♀) – or RV ejection fraction >40 – ≤45% | |
| II. Histopathology (endomyocardial biopsy) | ||
| Major | Residual myocytes <60% by morphometric analysis (or <50% if estimated), with fibrous replacement of the RV free wall myocardium ≥1 sample, with or without fatty replacement | |
| Minor | Residual myocytes 60–75% by morphometric analysis (or 50–65% if estimated), with fibrous replacement of the RV free wall ≥1 sample, with or without fatty replacement | |
| III. Repolarisation abnormalities (>14 years of age) | ||
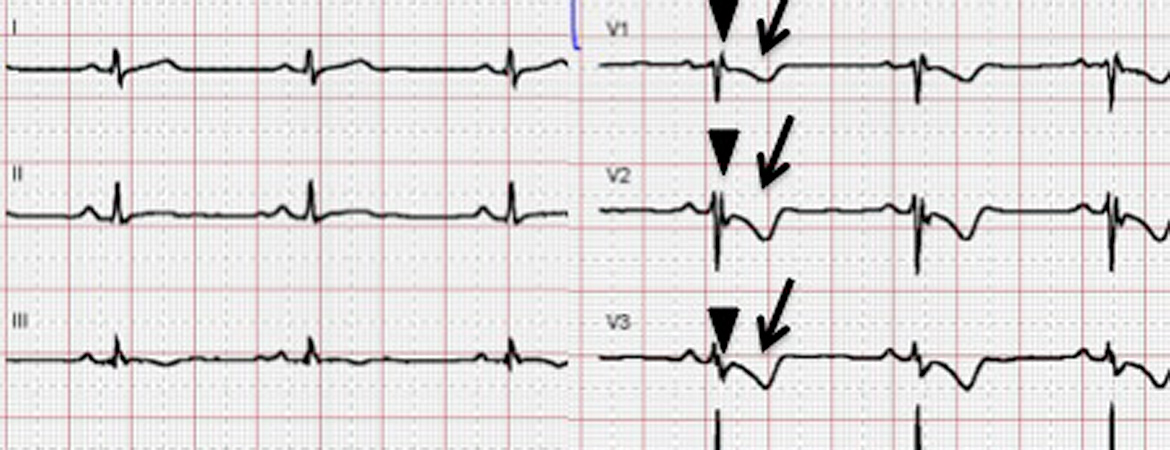
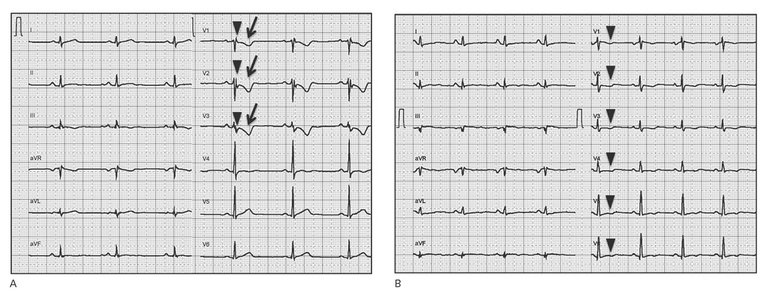
| Major | – T-wave inversions V1–V3 or beyond (in absence of complete RBBB) | |
| Minor | – T-wave inversions V1–V2 or V4–V6 (in absence of complete RBBB) – T-wave inversions V1–V4, if complete RBBB present | |
| IV. Depolarisation abnormalities | ||
| Major | Epsilon wave (reproducible low-amplitude signals between end of QRS complex to onset of the T-wave) in V1 to V3 | |
| Minor | Signal-averaged ECG (SAECG) with late potentials (if QRS on standard surface ECG <110 ms) | |
| V. Arrhythmias | ||
| Major | – Non-sustained or sustained ventricular tachycardia (VT) of LBBB morphology with superior axis | |
| Minor | – Non-sustained or sustained VT of LBBB morphology with inferior or unknown axis – >500 VES per 24 h (Holter) | |
| VI. Family history | ||
| Major | – ARVC in a first-degree relative who meets current Task Force Criteria – ARVC confirmed pathologically at autopsy or surgery in a first-degree relative – Identification of a pathogenic mutation categorised as associated with ARVC/D in index patient | |
| Minor | – Suspected ARVC in a first-degree relative (current Task Force criteria cannot be determined) – Premature SCD (<35 years of age) due to suspected ARVC in a first-degree relative – ARVC confirmed pathologically or by current Task Force Criteria in second-degree relatives | |
Published under the copyright license.
"Attribution - Non-Commercial - NoDerivatives 4.0"
No commercial reuse without permission.
See: emh.ch/en/emh/rights-and-licences/
